艾尼亚健康试管试管之家前言:肾结石和肾钙质沉着症的肾结石病,是一种患病率增加的临床综合征,具有显着的异质性。结石成分、表现年龄、复发率和肾功能损害因潜在病因而异。虽然钙基肾结石占绝大多数,但它们的病因仍然知之甚少。最近的研究强调了遗传易感性与饮食习惯一起构成肾结石形成的主要驱动因素的概念。除了在成年人群中最有可能被低估的单基因(孟德尔)疾病外,常见风险等位基因还解释了观察到的遗传性的一部分。有趣的是,已鉴定的GWAS位点通常与孟德尔病基因的位点相匹配,反之亦然(CASR,SLC34A1,CYP24A1)。这些发现提供了与肾钙稳态、维生素 D 代谢和 CaSR 信号传导相关的机制联系,这些信号传导由 CaSR–CLDN14–CLDN16/19 轴(副细胞 Ca)调节。2+重吸收)和TRPV5(跨细胞钙)2+ 重吸收)。最近鉴定了钙-草酸-肾结石(SLC26A1,CLDN2)和远端肾小管性酸中毒伴肾钙质沉着症(FOXI1,WDR72,ATP6V1C2)的新单基因疾病,从而进一步了解肾-肠轴和适当尿酸化的分子先决条件。实施关于遗传性肾结石疾病的集中患者登记是必要的,以便为迫切需要的临床研究建立特征良好的队列。
一、引言 - 肾结石形成的遗传性
由于11%的男性和7%的女性终生受肾结石影响,肾结石疾病的发病率和患病率在一般人群中继续上升[1]。肾结石及其实质形式的肾钙质沉着症统称为晶体病,因为它们都是由肾小管系统或其邻近的实质中的病理性晶体形成和沉淀引起的。晶体病是主要的健康负担,特别是在西方世界,与显著的发病率和进展为慢性肾脏疾病有关[1]。慢性肾脏疾病的风险最高,包括频繁复发、重复性手术/内镜干预以及伴有炎症。简单地说,肾结石的形成是由尿液中结晶抑制剂(例如柠檬酸盐,镁,尿路上腺素,焦磷酸盐)和启动子(例如草酸盐,钙,磷酸盐,尿酸盐,胱氨酸)的不平衡引起的,超过过饱和度,在Randall斑块处连续聚集,成核和结石生长(图1)。促进剂和/或抑制剂的肠内和/或肾脏稳态改变可导致失衡,例如肠内分泌不良(例如草酸盐)或肾脏吸收不良(例如磷酸盐/钙)(图1)。
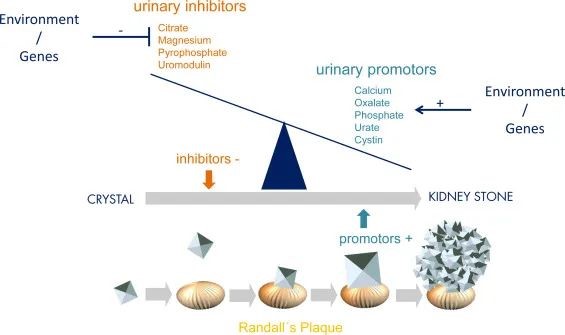
①图 1.尿液抑制剂和结晶促进剂的不平衡导致肾结石形成。尿液抑制剂和启动子的浓度受肠道和肾脏转运蛋白(胃肠道-肾轴)的影响和控制。这些转运蛋白和交换剂,如SLC26A1,负责分泌和吸收。关于草酸盐,肠内吸收过多但分泌不良,和/或肾分泌过多但吸收不良导致尿液草酸盐水平超过过饱和度,从而促进结晶和连续结石形成(改编自Pfau等人。 [68])。
②病因是多因素的,具有环境、显著的饮食、激素和遗传成分。为了估计总体遗传贡献,根据两项双胞胎研究的一致性率计算遗传性。在男性双胞胎中,肾结石的遗传性估计为56-57%,而女性双胞胎的首次数据表明遗传力显着降低(46%),这可能是观察到的肾结石总体发病率性别差异的原因[2],[3]。此外,多达 2/3 的高钙性结石成因患者有肾结石亲属[4]。由于超过80%的肾结石是钙基的,高钙尿症是肾结石形成者中占主导地位的代谢特征,因此其遗传因素一直在加强研究[5]。在不同人群中的全基因组关联研究(GWAS)和病例对照研究确定了多个相关的共同变异(SNP),其中度至小效应大小(OR 1.2-2.1)和候选基因的低外显率匹配位点,如ALPL(冰岛,日本,中国),CASR(冰岛,印度,台湾),CLDN14(冰岛/荷兰,冰岛,日本,印度,加拿大),SLC34A1(冰岛,日本),TRPV5 (冰岛、台湾)、TRPV6(瑞士)和 VDR(土耳其、意大利)[6]、[7]、[8]、[9]、[10]、[11]、[12]、[13]、[14]、[15]、[16]。相反,在冰岛和荷兰的病例中,已经发现UMD(rs4293393)的常见变异(rs4293393)对肾结石疾病具有保护作用(OR 0.88),可能是通过脲浑都胰岛素介导的TRPV5表面表达增强,这是一种高选择性Ca2+远端弯曲小管中的通道负责经细胞 Ca2+重吸收[17],[18]。另一方面,高钙尿症和钙结石形成是许多单基因疾病的一部分,其特征在于(超)大效应尺寸和高外显率的罕见变体(图2A)。值得注意的是,许多风险等位基因与已鉴定的单基因疾病共享其位点,例如在ALPL,CASR,SLC34A1,CYP24A1和SLC2A9中提供了晶体病中多基因和单基因遗传之间的联系[19],[20](图2A,表1)).在最近来自日本和英国的跨种族GWAS荟萃分析中,总共报告了20个肾结石相关位点。虽然大多数位点对先前的研究是确凿的,但另外7个先前未报告的位点被鉴定出来[21]。其中,发现了五个对钙敏感受体(CaSR)信号传导有预测影响的位点(DGKD,DGKH,WDR72,GPIC1和BCR),使该途径成为病理生理学考虑的中心。作为一种潜在的机制,高Ca激活CaSR2+- 和/或维生素D3的摄入,以及通过给予钙模拟物,被证明可以增加Henle厚厚的上行肢部的肾脏CLDN14表达,这反过来又抑制CLDN16 / 19介导的细胞旁钙2+重吸收[22],[23]。因此,所谓的CaSR-CLDN14/16/19轴失调被提出为高钙尿症的药学上适宜的靶标(图2B)[21]。
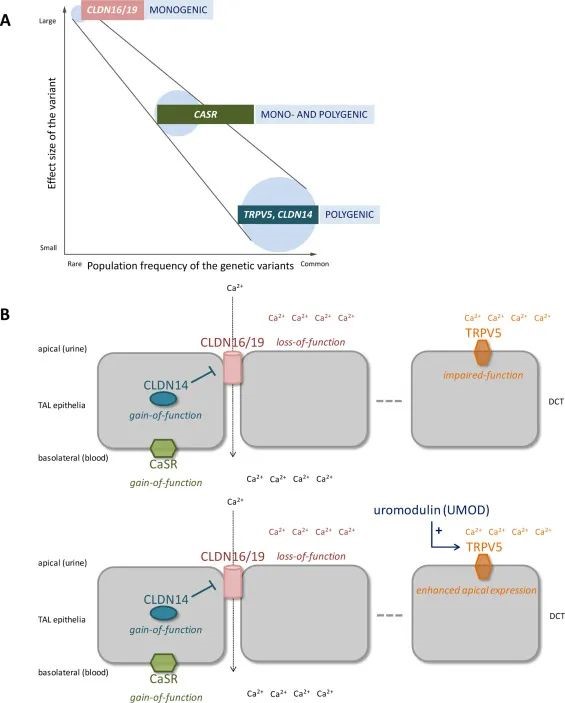
图 2.多基因在肾结石病中满足单基因遗传。高钙结石形成剂中遗传变异的效应大小,外显率和群体频率;单基因疾病(如家族性低镁血症伴高钙尿症和肾钙质沉着症 (FHHNC))(CLDN16/CLDN19)的特征是具有大效应量/高外显率的超细变异,但多基因疾病基于效应量小/外显率低但群体频率高的变异型 (A)。Henle(TAL)厚上行肢端的CaSR-CLDN14-CLDN16/19轴的简化模型,说明了共享机制通路中的单基因和多基因疾病(B)。钙尿症可能在CaSR和/或CLDN14功能增加时发生,导致细胞旁Ca抑制2+重吸收,类似于在FHHNC中看到的CLDN16/19的功能丧失(改编自Hou等人[69])。腔内尿球蛋白(UMOD)被认为通过增强顶端TRPV5表达和连续的跨细胞Ca来减少高钙尿症,从而具有保护作用 2+远端卷曲小管 (DCT) 的重吸收。
钙代谢:肾小管性酸中毒和肾钙质沉着症
腺苷酸环化酶10特发性(吸收性)高钙尿症,易感性143870常染色体显性遗传[40]
碱性磷酸酶,肝脏高钙尿症、肾钙质沉着症、低磷血症146300常染色体显性遗传/隐性[41]
ATP酶,H+转运,溶酶体V0亚基a4远端肾小管性酸中毒602722常染色体隐性遗传[42]
ATP酶,H+转运,溶酶体,V1亚基B1远端肾小管性酸中毒伴耳聋267300常染色体隐性遗传[43]
ATP酶,H+转运,溶酶体,V1亚基C2远端肾小管性酸中毒伴耳聋那常染色体隐性遗传[39]
碳酸酐酶 II石骨症 + 远端/近端肾小管性酸中毒259730常染色体隐性遗传[44]
钙感应受体低钙血症伴巴特综合征/低钙血症,常染色体显性遗传601198常染色体显性遗传[45]
氯离子通道,电压敏感型 5凹陷病/肾结石,1 型300009/310468X染色体隐性遗传[46]
氯化物通道,电压敏感型 Kb巴特综合征,3型607364常染色体隐性遗传[47]
Claudin 2特发性高钙尿症媒体报道中
Claudin 16家族性低镁血症伴高钙尿症和肾钙质沉着症248250常染色体隐性遗传[48]
Claudin 19家族性低镁血症伴高钙尿症和肾钙质沉着症伴眼部异常248190常染色体隐性遗传[49]
细胞色素P450,家族24,亚家族A,多肽11,25-(OH) D-24 羟化酶缺乏症,婴儿高钙血症143880常染色体隐性遗传[50]
序列相似性家族 20,成员 A牙釉质肾综合征、无色病不全和肾钙质沉着症204690常染色体隐性遗传[51]
叉头盒蛋白I1肾钙质沉着症、远端肾小管性酸中毒、前庭导水管增大600791常染色体隐性遗传[36]
肝细胞核因子4,αMODY + 范可尼综合征 + 肾钙质沉着症 (p.R76 W)125850常染色体显性遗传[52]
钾向内整流通道,J亚科,成员1巴特综合征,2型241200常染色体隐性遗传[53]
黑色素瘤抗原,家族D,2巴特综合征,5型300971X染色体隐性遗传[54]
Lowe的眼球肾综合征洛威综合征/凹痕病 2309000/300555X染色体隐性遗传[55]
溶质载体家族 12,成员 1巴特综合征,1型601678常染色体隐性遗传[56]
溶质载体家族4,阴离子交换剂,成员1原发性远端肾小管性酸中毒,显性/隐性179800/611590常染色体显性遗传/隐性[57]
维生素D(1,25-二羟基维生素D3)受体特发性高钙尿症277440常染色体显性遗传[58]
含蛋白质72的WD重复序列肾钙质沉着症、远端肾小管性酸中毒、发育不全、IIA3 型613211常染色体隐性遗传[38]
草酸盐代谢
丙氨酸-乙醛酸氨基转移酶原发性高草酸尿症,1 型259900常染色体隐性遗传[26]
乙醛酸还原酶/羟基丙酮酸还原酶原发性高草酸尿症,2 型260000常染色体隐性遗传[27]
4-羟基-2-氧代戊二酸醛缩酶1原发性高草酸尿症,3 型613616常染色体隐性遗传[28]
溶质载体家族26(硫酸盐转运蛋白),成员1草酸钙肾结石167030常染色体隐性遗传[34]
胱氨酸尿症
溶质载体家族3(胱氨酸,二元和中性氨基酸转运蛋白,胱氨酸激活剂,双基和中性氨基酸转运),成员1胱氨酸尿症,A型220100常染色体隐性遗传[59]
溶质载体家族7(糖蛋白相关氨基酸转运蛋白轻链,bo,+系统),成员9胱氨酸尿症,B型220100常染色体显性遗传/隐性[60]
尿酸结石
次黄嘌呤磷酸核糖基转移酶1Kelley-Seegmiller 综合征、部分次黄嘌呤磷酸核糖基转移酶缺乏、次黄嘌呤磷酸核糖基转移酶相关性痛风300323X染色体隐性遗传[61]
溶质载体家族22(有机阴离子/尿酸盐转运蛋白),成员12肾低尿酸血症,1 型220150常染色体显性遗传/隐性[62]
溶质载体家族2(促进葡萄糖转运蛋白),成员9肾低尿酸血症,2 型612076常染色体显性遗传/隐性[20]
肾脏磷酸盐消耗
溶质载体家族34(磷酸钠),成员1低血吸性肾结石、骨质疏松症-1/范可尼肾结核综合征 2612286/613388常染色体显性遗传/隐性[63]
溶质载体家族34(磷酸钠),成员3低相噬细胞性佝偻病伴高钙尿症241530常染色体隐性遗传[64]
溶质载体家族9,亚科A(NHE3,阳离子质子反携带者3),成员3调节器1低血吸虫肾结石,骨质疏松症-2612287常染色体显性遗传[65]
其他代谢紊乱
腺嘌呤磷酸核糖基转移酶腺嘌呤磷酸核糖基转移酶缺乏症614723常染色体隐性遗传[66]
黄嘌呤脱氢酶黄素尿,1型278300常染色体隐性遗传[67]
MIM:孟德尔在人类中的继承;MODY:年轻人的成熟期糖尿病。
a在肾结石疾病的全基因组关联研究中也被发现为候选位点的基因。
b对于HNF4A,MIM表型数表示MODY 1型,因为Fanconi综合征和肾钙质沉着症的发生仅在存在特定等位基因时才显示:p.R76W。
迄今为止,在晶体病患者中已经鉴定出近40种具有在线人类孟德尔遗传(OMIM)定义表型的单基因疾病(表1)。与所有孟德尔疾病一样,这些单基因形式以常染色体显性遗传、常染色体隐性遗传或 X 连锁方式传播。有趣的是,在其中几种疾病中,已经报道了隐性和显性遗传模式:ALPL,CYP24A1,SLC7A9,SLC34A1,SLC34A3,SLC2A9,SLC22A12和SLC4A1,指向基因剂量效应。例如,在编码肾近端小管(NaPi2c)中主要磷酸转运蛋白之一的SLC34A3中,表明杂合子个体不再仅仅被视为健康携带者,因为它们显示肾钙化和/或骨骼表现明显比野生型个体更频繁;但仍然低于双合子(纯合子, 复合杂合子)个体[24]。据报道,CYP24A1突变的家族也有类似的观察结果[25]。虽然大多数综合征和重度先天性疾病表现出隐性遗传模式(Bartter,Lowe,Dent,FHHNC,远端肾小管性酸中毒伴感音神经性耳聋),但较轻的疾病与主要传播遗传性疾病的突变有关。从机理上讲,大多数编码蛋白构成肾溶质转运蛋白(例如SLC34A1 / NaPi2a,SLC34A3 / NaPi2c,SLC9A3R1),氯化物通道(CLCN5),紧密连接蛋白(例如CLDN16 / CLDN19)和代谢酶(例如AGXT,APRT,CYP24A1)。因此,潜在的缺陷主要位于肾脏本身的肾小管系统中,因此可归因于肾小管病。相反,肾外疾病,如原发性高草酸尿症,其中肝酶功能障碍(AGXT,GRHPR,HOGA1)导致草酸盐积累并伴有继发性肾病,也是晶体病的可想象原因[26],[27],[28]。虽然每种疾病表型被认为代表了一种相对罕见的实体,但多种单基因病因可能通过其广泛的遗传异质性共同解释大量患者。最近研究了单基因疾病对肾结石疾病总体患病率的贡献。虽然基于结石成分分析,先前得出结论,儿童单基因病因不超过9.6%,成人不超过1.6%,但3项遗传学研究发现,儿童的贡献率高达30%,成人为11%[29],[30],[31],[32],[33]。在第一项研究中,来自典型肾结石诊所的268个基因未解决个体的队列;通过基因组合方法对102名儿科和166名成年先带进行了分析,以分析30种晶体病单基因疾病的罕见变异,导致15%的病例得到分子诊断。在儿科亚组中,诊断率为21%,而在成人中,有害变异存在于11%[30]。两项随访研究能够在儿科和青少年队列中证实这些结果。首先,在一个由143名晶体病患者组成的儿科队列中,17%的病例由14个不同基因的致病变异解释[31]。其次,在51个年龄在25岁之前的晶体显现的家族的队列中,靶向WES用于检测近30%的遗传原因[32]。毫不奇怪,隐性突变在新生儿和先天性疾病病例中更常见,而显性疾病通常在晚年表现出来。尽管由于在三级保健中心招募而对潜在的选择偏差存在争议,但这些数据累积表明遗传性肾结石疾病是一种未被充分诊断的疾病,特别是在年轻人中。
二、肾结石和肾钙质沉着症新疾病基因的鉴定
除了测试已知孟德尔疾病的患病率外,下一代测序技术还推动了晶体病的基因鉴定领域。最近的一项新发现与SLC26A1中人类突变的发现有关[34]。自2010年Dawson等人首次描述Slc26a1(Sat1)-敲除小鼠中的钙-草酸盐肾结石形成和肾钙质沉着症以来,SLC26A1一直是真正的肾结石候选基因[35]。SLC26A1 编码在近端肾小管、回肠和空肠的基底外侧膜上表达的阴离子交换剂。因此,通过使用候选基因方法,在具有早发性钙-草酸肾结石病史的人类中鉴定出致病变异,即两个具有双巩错义变异的无关个体[34]。在功能上,已鉴定变异的致病性在体外被证明,导致细胞内脱斜和转运活性受损[34]。因此,有缺陷的SLC26A1提供了钙-草酸肾结石的新机制,在测试个体复发性钙-草酸盐结石形成的遗传原因时应考虑这一点。同样,CLDN2(claudin 2)中的遗传变异编码细胞旁阳离子通道,介导近端小管的钙重吸收,与小鼠和人类的高钙尿有关。常见和罕见的CLDN2变异都与钙肾结石和肾钙质沉着症有关,无论是在遗传敏感性方面还是通过X连锁隐性遗传。
在远端肾小管性酸中毒和伴随肾钙质沉着症领域,近年来已经表征了三种新的遗传参与者(FOXI1,WDR72,ATP6V1C2)。在编码叉头转录因子的FOXI1中,双性遗传变异最近与早发性远端肾小管性酸中毒、肾钙质沉着症和感音神经性耳聋有关[36]。此外,WDR72先前与V型质子ATP酶运输有关,在远端肾小管性酸中毒伴或不伴有无发育不全性的患者中发现变异。类似地,在远端肾小管性酸中毒的遗传性未解决病例中,全外显子组测序最近导致鉴定出ATP6V1C2的双列功能丧失变体,该变体编码在肾收集管中插入细胞的顶端膜上表达的V型质子ATP酶的另一个亚基,这对于适当的尿液酸化过程至关重要]。
三、遗传性肾结石疾病需要集中患者登记
在美国,罕见肾结石联盟(RKSC)构成了一个平台,整合和协调了胱氨酸尿症,原发性高草酸尿症,腺嘌呤磷酸核糖基转移酶缺乏症,Dent和Lowe病等罕见病症的注册,基础科学和临床研究活动,没有集中收集遗传性肾结石患者的临床和遗传数据收集 截至今天,疾病已在欧洲实施。我们与现有的欧洲原发性高草酸血症登记处OxalEurope(教授.B. Hoppe)合作,并通过Deutsche Forschungsgemeinschaft(DFG)和Else Kröner-Fresenius Stiftung(EKFS)的资助,我们在德国莱比锡大学建立了临床患者“遗传性肾结石疾病登记处”。集中式登记对于建立特征良好的患者队列以供未来的临床研究非常重要。在欧洲,欧洲罕见肾脏疾病参考网络已开始提供一个组织平台,以集中各种子注册和连接不同形式的遗传性肾结石疾病的数据(ERKReg)。
艾尼亚健康试管试管婴儿平台小Tips:希望这些努力将显着提高我们的病理生理学理解,并帮助启动临床研究,最终为频繁复发的肾结石形成者提供更个性化的预防和治疗。
HSG方便、廉价,可以检查输卵管近端和远端的阻塞,显示峡部的结节性输卵管炎,了解输卵管的细节并评估输卵管周围的炎症情况。
IVF实验室的胚胎学家在取卵前应与临床医生核对患者信息,在临床医师对患者实施取卵术时,应该同步进行捡卵操作。
PGD过程中的胚胎活检是一种创伤性显微操作,有可能影响活检后胚胎发育能力。在进行胚胎活检之前需要在透明带上打孔或者部…
胚胎冷冻保存对FET妊娠率以及子代出生缺陷率没有显著影响。在液氮中细胞的酶活力几乎完全被抑制,细胞代谢过程处于停滞状…
胚胎冷冻保存技术是辅助生殖技术(ART)重要的衍生技术之一,是指将胚胎置于超低温环境(液氮,-196℃)中冷冻保存,…







